
quinta-feira, 6 de março de 2014

sábado, 15 de fevereiro de 2014
quinta-feira, 13 de fevereiro de 2014
domingo, 24 de novembro de 2013

Nosso objetivo é estudar os microrganismos presentes no Bioma Caatinga e de que forma podem auxiliar no desenvolvimento agrícola das regiões semiáridas.
Também estaremos desenvolvendo trabalhos ligados a recuperação de áreas degradadas.
domingo, 27 de janeiro de 2013
Retomando as Postagens!!!!
Pessoal,
Novo ano começando, e com ele mais um semestre letivo também.
Voltarei a postar periodicamente informações recentes sobre Biotecnologia e Microbiologia.
A dica de hoje é o livro lançado em 2012 pela Sociedade Brasileira de Genética, de Autoria da Drª Marillis do Valle Marques, o livro é intitulado: Biologia Molecular e Genética Bacteriana.
A Drª Marillis possui doutorado em Bioquímica pela Universidade de São Paulo e pós-Doutorado em Biologia molecular pela University of California - Los Angeles (UCLA).
O livro apresenta uma diagramação muito boa, ótimas figuras, linguagem muito clara e objetiva, além de possuir um conteudo atual e moderno.
Indico o livro para todos os alunos das Ciências Biológicas que pegam a disciplina de Biologia Molecular bem como para aqueles que pegam Microbiologia.
 Aos meus alunos de Microbiologia, este será nosso livro texto para o assunto: Genética Bacteriana.
Aos meus alunos de Microbiologia, este será nosso livro texto para o assunto: Genética Bacteriana.
Bom Ano à todos!!!!!
Adailson Feitoza
Novo ano começando, e com ele mais um semestre letivo também.
Voltarei a postar periodicamente informações recentes sobre Biotecnologia e Microbiologia.
A dica de hoje é o livro lançado em 2012 pela Sociedade Brasileira de Genética, de Autoria da Drª Marillis do Valle Marques, o livro é intitulado: Biologia Molecular e Genética Bacteriana.
A Drª Marillis possui doutorado em Bioquímica pela Universidade de São Paulo e pós-Doutorado em Biologia molecular pela University of California - Los Angeles (UCLA).
O livro apresenta uma diagramação muito boa, ótimas figuras, linguagem muito clara e objetiva, além de possuir um conteudo atual e moderno.
Indico o livro para todos os alunos das Ciências Biológicas que pegam a disciplina de Biologia Molecular bem como para aqueles que pegam Microbiologia.
Bom Ano à todos!!!!!
Adailson Feitoza
quarta-feira, 19 de setembro de 2012
ENCODE: The human encyclopaedia
ENCODE: The human encyclopaedia
First they sequenced it. Now they have surveyed its hinterlands.
But no one knows how much more information the human genome holds, or
when to stop looking for it.

ENCODE was designed to pick up where the Human Genome Project left off. Although that massive effort revealed the blueprint of human biology, it quickly became clear that the instruction manual for reading the blueprint was sketchy at best. Researchers could identify in its 3 billion letters many of the regions that code for proteins, but those make up little more than 1% of the genome, contained in around 20,000 genes — a few familiar objects in an otherwise stark and unrecognizable landscape. Many biologists suspected that the information responsible for the wondrous complexity of humans lay somewhere in the ‘deserts’ between the genes. ENCODE, which started in 2003, is a massive data-collection effort designed to populate this terrain. The aim is to catalogue the ‘functional’ DNA sequences that lurk there, learn when and in which cells they are active and trace their effects on how the genome is packaged, regulated and read.
After an initial pilot phase, ENCODE scientists started applying their methods to the entire genome in 2007. Now that phase has come to a close, signalled by the publication of 30 papers, in Nature, Genome Research and Genome Biology. The consortium has assigned some sort of function to roughly 80% of the genome, including more than 70,000 ‘promoter’ regions — the sites, just upstream of genes, where proteins bind to control gene expression — and nearly 400,000 ‘enhancer’ regions that regulate expression of distant genes (see page 57)1. But the job is far from done, says Birney, a computational biologist at the European Molecular Biology Laboratory’s European Bioinformatics Institute in Hinxton, UK, who coordinated the data analysis for ENCODE. He says that some of the mapping efforts are about halfway to completion, and that deeper characterization of everything the genome is doing is probably only 10% finished. A third phase, now getting under way, will fill out the human instruction manual and provide much more detail.
Many who have dipped a cup into the vast stream of data are excited by the prospect. ENCODE has already illuminated some of the genome’s dark corners, creating opportunities to understand how genetic variations affect human traits and diseases. Exploring the myriad regulatory elements revealed by the project and comparing their sequences with those from other mammals promises to reshape scientists’ understanding of how humans evolved.
Yet some researchers wonder at what point enough will be enough. “I don’t see the runaway train stopping soon,” says Chris Ponting, a computational biologist at the University of Oxford, UK. Although Ponting is supportive of the project’s goals, he does question whether some aspects of ENCODE will provide a return on the investment, which is estimated to have exceeded US$185 million. But Job Dekker, an ENCODE group leader at the University of Massachusetts Medical School in Worcester, says that realizing ENCODE’s potential will require some patience. “It sometimes takes you a long time to know how much can you learn from any given data set,” he says.
Even before the human genome sequence was finished2, the National Human Genome Research Institute (NHGRI), the main US funder of genomic science, was arguing for a systematic approach to identify functional pieces of DNA. In 2003, it invited biologists to propose pilot projects that would accrue such information on just 1% of the genome, and help to determine which experimental techniques were likely to work best on the whole thing.
Nature Podcast
ENCODE coordinator Ewan Birney discusses consortium science with the LHC’s Tejinder Virdee.The 32 groups, including more than 440 scientists, focused on 24 standard types of experiment (see ‘Making a genome manual’). They isolated and sequenced the RNA transcribed from the genome, and identified the DNA binding sites for about 120 transcription factors. They mapped the regions of the genome that were carpeted by methyl chemical groups, which generally indicate areas in which genes are silent. They examined patterns of chemical modifications made to histone proteins, which help to package DNA into chromosomes and can signal regions where gene expression is boosted or suppressed. And even though the genome is the same in most human cells, how it is used is not. So the teams did these experiments on multiple cell types — at least 147 — resulting in the 1,648 experiments that ENCODE reports on this week1, 4–8.
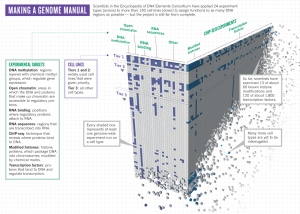
The real fun starts when the various data sets are layered together. Experiments looking at histone modifications, for example, reveal patterns that correspond with the borders of the DNaseI-sensitive sites. Then researchers can add data showing exactly which transcription factors bind where, and when. The vast desert regions have now been populated with hundreds of thousands of features that contribute to gene regulation. And every cell type uses different combinations and permutations of these features to generate its unique biology. This richness helps to explain how relatively few protein-coding genes can provide the biological complexity necessary to grow and run a human being. ENCODE “is much more than the sum of the parts”, says Manolis Kellis, a computational genomicist at the Massachusetts Institute of Technology in Cambridge, who led some of the data-analysis efforts.
The data, which have been released throughout the project, are already helping researchers to make sense of disease genetics. Since 2005, genome-wide association studies (GWAS) have spat out thousands of points on the genome in which a single-letter difference, or variant, seems to be associated with disease risk. But almost 90% of these variants fall outside protein-coding genes, so researchers have little clue as to how they might cause or influence disease.
The map created by ENCODE reveals that many of the disease-linked regions include enhancers or other functional sequences. And cell type is important. Kellis’s group looked at some of the variants that are strongly associated with systemic lupus erythematosus, a disease in which the immune system attacks the body’s own tissues. The team noticed that the variants identified in GWAS tended to be in regulatory regions of the genome that were active in an immune-cell line, but not necessarily in other types of cell and Kellis’s postdoc Lucas Ward has created a web portal called HaploReg, which allows researchers to screen variants identified in GWAS against ENCODE data in a systematic way. “We are now, thanks to ENCODE, able to attack much more complex diseases,” Kellis says.
Are we there yet?
Researchers could spend years just working with ENCODE’s existing data — but there is still much more to come. On its website, the University of California, Santa Cruz, has a telling visual representation of ENCODE’s progress: a grid showing which of the 24 experiment types have been done and which of the nearly 180 cell types ENCODE has now examined. It is sparsely populated. A handful of cell lines, including the lab workhorses called HeLa and GM12878, are fairly well filled out. Many, however, have seen just one experiment.Scientists will fill in many of the blanks as part of the third phase, which Birney refers to as the ‘build out’. But they also plan to add more experiments and cell types. One way to do that is to expand the use of a technique known as chromatin immunoprecipitation (ChIP), which looks for all sequences bound to a specific protein, including transcription factors and modified histones. Through a painstaking process, researchers develop antibodies for these DNA binding proteins one by one, use those antibodies to pull the protein and any attached DNA out of cell extracts, and then sequence that DNA.
But at least that is a bounded problem, says Birney, because there are thought to be only about 2,000 such proteins to explore. (ENCODE has already sampled about one-tenth of these.) More difficult is figuring out how many cell lines to interrogate. Most of the experiments so far have been performed on lines that grow readily in culture but have unnatural properties. The cell line GM12878, for example, was created from blood cells using a virus that drives the cells to reproduce, and histones or other factors may bind abnormally to its amped-up genome. HeLa was established from a cervical-cancer biopsy more than 50 years ago and is riddled with genomic rearrangements. Birney recently quipped at a talk that it qualifies as a new species.
ENCODE researchers now want to look at cells taken directly from a person. But because many of these cells do not divide in culture, experiments have to be performed on only a small amount of DNA, and some tissues, such as those in the brain, are difficult to sample. ENCODE collaborators are also starting to talk about delving deeper into how variation between people affects the activity of regulatory elements in the genome. “At some places there’s going to be some sequence variation that means a transcription factor is not going to bind here the same way it binds over here,” says Mark Gerstein, a computational biologist at Yale University in New Haven, Connecticut, who helped to design the data architecture for ENCODE. Eventually, researchers could end up looking at samples from dozens to hundreds of people.
The range of experiments is expanding, too. One quickly developing area of study involves looking at interactions between parts of the genome in three-dimensional space. If the intervening DNA loops out of the way, enhancer elements can regulate genes hundreds of thousands of base pairs away, so proteins bound to the enhancer can end up interacting with those attached near the gene. Dekker and his collaborators have been developing a technique to map these interactions. First, they use chemicals that fuse DNA-binding proteins together. Then they cut out the intervening loops and sequence the bound DNA, revealing the distant relationships between regulatory elements. They are now scaling up these efforts to explore the interactions across the genome. “This is beyond the simple annotation of the genome. It’s the next phase,” Dekker says.
The question is, where to stop? Kellis says that some experimental approaches could hit saturation points: if the rate of discoveries falls below a certain threshold, the return on each experiment could become too low to pursue. And, says Kellis, scientists could eventually accumulate enough data to predict the function of unexplored sequences. This process, called imputation, has long been a goal for genome annotation. “I think there’s going to be a phase transition where sometimes imputation is going to be more powerful and more accurate than actually doing the experiments,” Kellis says.
Yet with thousands of cell types to test and a growing set of tools with which to test them, the project could unfold endlessly. “We’re far from finished,” says geneticist Rick Myers of the HudsonAlpha Institute for Biotechnology in Huntsville, Alabama. “You might argue that this could go on forever.” And that worries some people. The pilot ENCODE project cost an estimated $55 million; the scale-up was about $130 million; and the NHGRI could award up to $123 million in the next phase.
Some researchers argue that they have yet to see a solid return on that investment. For one thing, it has been difficult to collect detailed information on how the ENCODE data are being used. Mike Pazin, a programme director at the NHGRI, has scoured the literature for papers in which ENCODE data played a significant part. He has counted about 300, 110 of which come from labs without ENCODE funding. The exercise was complicated, however, because the word ‘encode’ shows up in genetics and genomics papers all the time. “Note to self,” says Pazin wryly, “make up a unique project name next time around.”
A few scientists contacted for this story complain that this isn’t much to show from nearly a decade of work, and that the choices of cell lines and transcription factors have been somewhat arbitrary. Some also think that the money eaten up by the project would be better spent on investigator-initiated, hypothesis-driven projects — a complaint that also arose during the Human Genome Project. But unlike the genome project, which had a clear endpoint, critics say that ENCODE could continue to expand and is essentially unfinishable. (None of the scientists would comment on the record, however, for fear that it would affect their funding or that of their postdocs and graduate students.)
Birney sympathizes with the concern that hypothesis-led research needs more funding, but says that “it’s the wrong approach to put these things up as direct competition”. The NHGRI devotes a lot of its research dollars to big, consortium-led projects such as ENCODE, but it gets just 2% of the total US National Institutes of Health budget, leaving plenty for hypothesis-led work. And Birney argues that the project’s systematic approach will pay dividends. “As mundane as these cataloguing efforts are, you’ve got to put all the parts down on the table before putting it together,” he says.
After all, says Gerstein, it took more than half a century to get from the realization that DNA is the hereditary material of life to the sequence of the human genome. “You could almost imagine that the scientific programme for the next century is really understanding that sequence.”
FONTE: http://www.nature.com/news/encode-the-human-encyclopaedia-1.11312
Assinar:
Postagens (Atom)